Химия - Теория функционала плотности - Формальное обоснование метода
01 марта 2011Оглавление:
1. Теория функционала плотности
2. Описание метода
3. Формальное обоснование метода
4. Приближения
5. Обобщение на случай магнитного поля
6. Применения
7. Программное обеспечение, которое реализует метод теории функционала плотности
Согласно приближению Борна — Оппенгеймера, которое применяется в большинстве расчётов электронной структуры, ядра, входящие в состав рассматриваемой системы, считаются неподвижными. Электростатический потенциал V, создаваемый этими «неподвижными» ядрами, является внешним для электронов. Стационарное состояние электронов описывается волновой функцией  , которая является решением уравнения Шрёдингера
, которая является решением уравнения Шрёдингера
где H — гамильтониан электронной подсистемы, N — количество электронов, U описывает электрон-электронное взаимодействие. Операторы T и U одинаковы для всех систем, в то время как вид V зависит от конкретной системы. Как видно, основное отличие одночастичной задачи от задачи многих тел состоит в наличии слагаемого, описывающего электрон-электронное взаимодействие, U. Существует большое количество методов решения многочастичного уравнения Шрёдингера, основанных на разложении волновой функции с использованием определителя Слэтера. Простейший из них — метод Хартри — Фока, на основе которого развит ряд современных методов. Общей проблемой для них является значительная вычислительная трудоёмкость, из-за которой область применения метода Хартри — Фока и производных от него ограничена не слишком большими системами.
Метод теории функционала плотности в значительной степени решает проблему расчёта систем, включающих большое число частиц, путём сведения задачи о системе многих тел с потенциалом электрон-электронного взаимодействия U к одночастичной задаче, в которой слагаемое U отсутствует.
Плотность частиц,  , с помощью которой и строится формализм теории функционала плотности, задается выражением:
, с помощью которой и строится формализм теории функционала плотности, задается выражением:

Хоэнберг и Кон в 1964 показали, что это выражение может быть обращено: по заданной плотности частиц в основном состоянии,  , можно найти соответствующую волновую функцию основного состояния
, можно найти соответствующую волновую функцию основного состояния  . Иными словами, Ψ0 — единственный функционал от n0, то есть
. Иными словами, Ψ0 — единственный функционал от n0, то есть
- Ψ0 = Ψ0,
а, следовательно, все остальные наблюдаемые физические величины O также являются функционалами n0:

В частности, для энергии основного состояния можно записать

где вклад внешнего потенциала  может быть переписан через плотность частиц:
может быть переписан через плотность частиц:
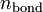
Функционалы T и U одинаковы для всех систем, а V, очевидно, зависит от вида рассматриваемой системы. Для заданной системы вид V известен, и можно минимизировать функционал
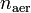
относительно распределения плотности частиц , если, конечно, имеются выражения для T и U. В результате минимизации получается плотность частиц в основном состоянии n0, а вместе с ней и все наблюдаемые в основном состоянии величины.
Вариационная задача отыскания минимума функционала энергии E может быть решена с помощью метода множителей Лагранжа, как это и было сделано Коном и Шэмом в 1965 г. Таким образом, функционал энергии в приведённом выше выражении может быть записан как эффективный функционал плотности частиц в одночастичной системе:
где Ts означает кинетическую энергию свободной частицы, а Vs — эффективный внешний потенциал для электронной подсистемы. Ясно, что  если Vs взят в виде
если Vs взят в виде
- Vs = V + U +.
Решение так называемых уравнений Кона — Шэма для вспомогательной системы, из которой исключено электрон-электронное взаимодействие,
даёт орбитали 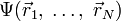 , по которым восстанавливается электронная плотность исходной многочастичной системы:
, по которым восстанавливается электронная плотность исходной многочастичной системы:
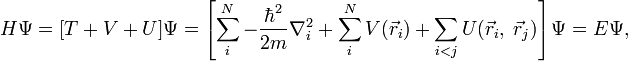
Эффективный одночастичный потенциал Vs записывается как
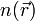
где второе слагаемое — слагаемое Хартри — описывает электрон-электронное кулоновское отталкивание, а последнее слагаемое VXC называется обменно-корреляционным потенциалом. Здесь VXC включает все многочастичные взаимодействия.
Поскольку слагаемое Хартри и член VXC зависят от плотности , которая зависит от , которая, в свою очередь, зависит от Vs, решение самосогласованных уравнений Кона — Шэма может быть произведено с помощью итеративной процедуры последовательных приближений. Как правило, отталкиваясь от начального приближения для , рассчитывается соответствующее слагаемое Vs, для которого затем решаются уравнения Кона — Шэма, из которых получается . Отсюда можно получить следующее приближение для плотности и т. д.
Просмотров: 10780
|
|